CRUST
R package for clonal deconvolution
This package is created to offer a consolidated tool for clonal deconvolution while using bulk sequencing data when a tumor is multiregionally or multi-temporally sampled. The package addresses several shortcomings while subclonally reconstructing several samples together that show up quite often due to either sample quality, processing, sequencing or inferential artifacts.
Installation
CRUST borrows clustering programs and other supporting facades from several other dependencies. Most of which has a continuing support from CRAN. Although more often than not one might find unable to import one or more dependencies due to lack of support from its maintainer. If such consern arises please refer to the package manual to find a list of direct and suggested dependencies which may help to resolve the issue.
install.packages("remotes")
remotes::install_github("ShixiangWang/copynumber")
remotes::install_github("Subhayan18/CRUST")
If no error message has popped up, we are now ready to load the package in global environment.
require(CRUST)
Preliminary usage
CRUST comes with one simulated tetraploid tumor data that has eight different samples. This is mostly used for demonstration purpose. We will use this data to perform our first clonal deconvolution. A set of example user input is given below but there are lot of other choices in methods and inputs that can be provided.
The cluster.doc function needs to be called for the analysis which requires the user to declare the colum numbers of the sample names, variant allele frequencies and among other options, a method to select the optimum number of clusters and a method to perform clustering. The last two options have a default input if left undeclared.
As the variant allele frequency or VAF is the currency to any deconvolution program, it is paramount to ensure its integrity prior to analysis. A major concern is thus to adjust the VAFs according to the segmental copy numbers of each sample. To this end CRUST only analyzes segments that have same allelic makeup across samples. For example, in our imaginary tetraploid tumor the allelic makeup can assume the following states:
0+4 : 0 mutatant and 4 wildtypes
1+3 : 1 mutatant and 3 wildtypes
2+2 : 2 mutatant and 2 wildtypes
3+1 : 3 mutatant and 1 wildtypes
4+0 : 4 mutatant and 0 wildtypes
Instead of adjusting the VAFs corresponding to their respective segmental copy numbers CRUST analyzes each distinct allelic composition in separate runs. The user is prompted to declare this allelic make up in the beginning of each run. In addition to this CRUST also asks the user to make a visual inspection of the VAF dot plot and provide a intuitive solution to the problem. It does not uses this intuitive solution to infer cluster separation rather builds upon this and later in the analyses provides the user with options to modify solutions.
## let's have a look at the data itself
?test.dat
head(test.dat)
r.es <- cluster.doc(test.dat, sample = 1, vaf = 2,
optimization.method = 'GMM', clustering.method = 'hkm')
## example user input:
## suspected chromosomal segmentation profile of the sample: 3+1
## How many clonal VAF clouds do you think are present: 2
## How many sub-clonal VAF clouds do you think are present: 2
## Would you like to see my suggestion instead? (yes/no): no
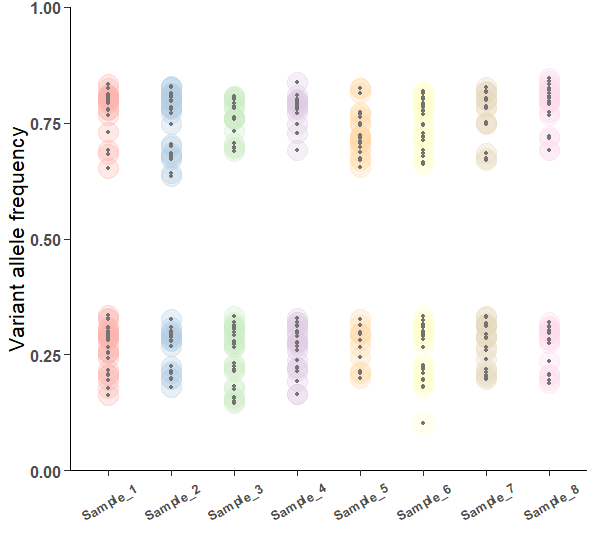
Figure 1 shows how the distribution of variant allele frequencies look for the 8 simulated samples. It is clear that two different clouds (lack of a better word) of VAFs exist centering approximately around 0.25 and 0.75. Depending on this structure of the spread it is concievable that the allelic segmentation is a tetraploid 3+1 with each cloud harboring one clonal and one subclonal clusters.
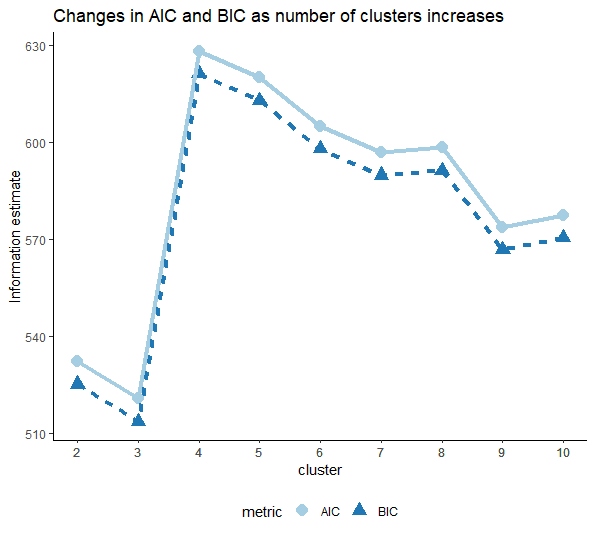
Figure 2 shows the changes in Bayesian Information criteria (BIC) and Akaike Information criterion (AIC) estimated based on the number of clusters fitted.
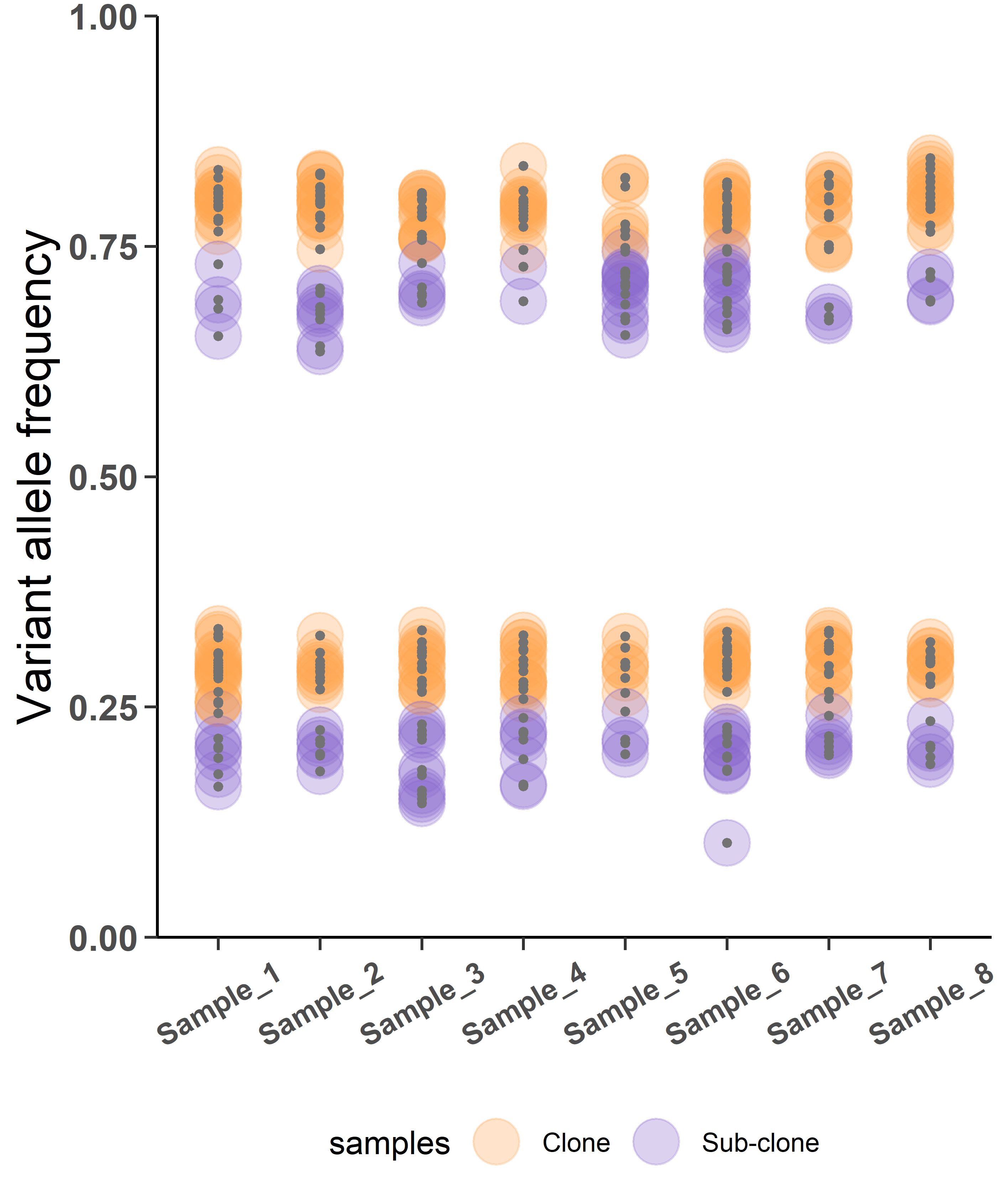
Figure 3 shows the clustered samples here depict the distribution of clonal and sub-clonal variants.
Scaling
Quite often the sequencing would not give as clear a picture as is seen here with the hypothetical data. Most likely the tumor samples will have normal cells mixed with the tumor cells, sometimes along with necrotic tissue. This affects the relative proportion of mutants in a sample. To account for changes in VAF due to varying sample purity CRUST scales the data whenever estimates of purity is provided.
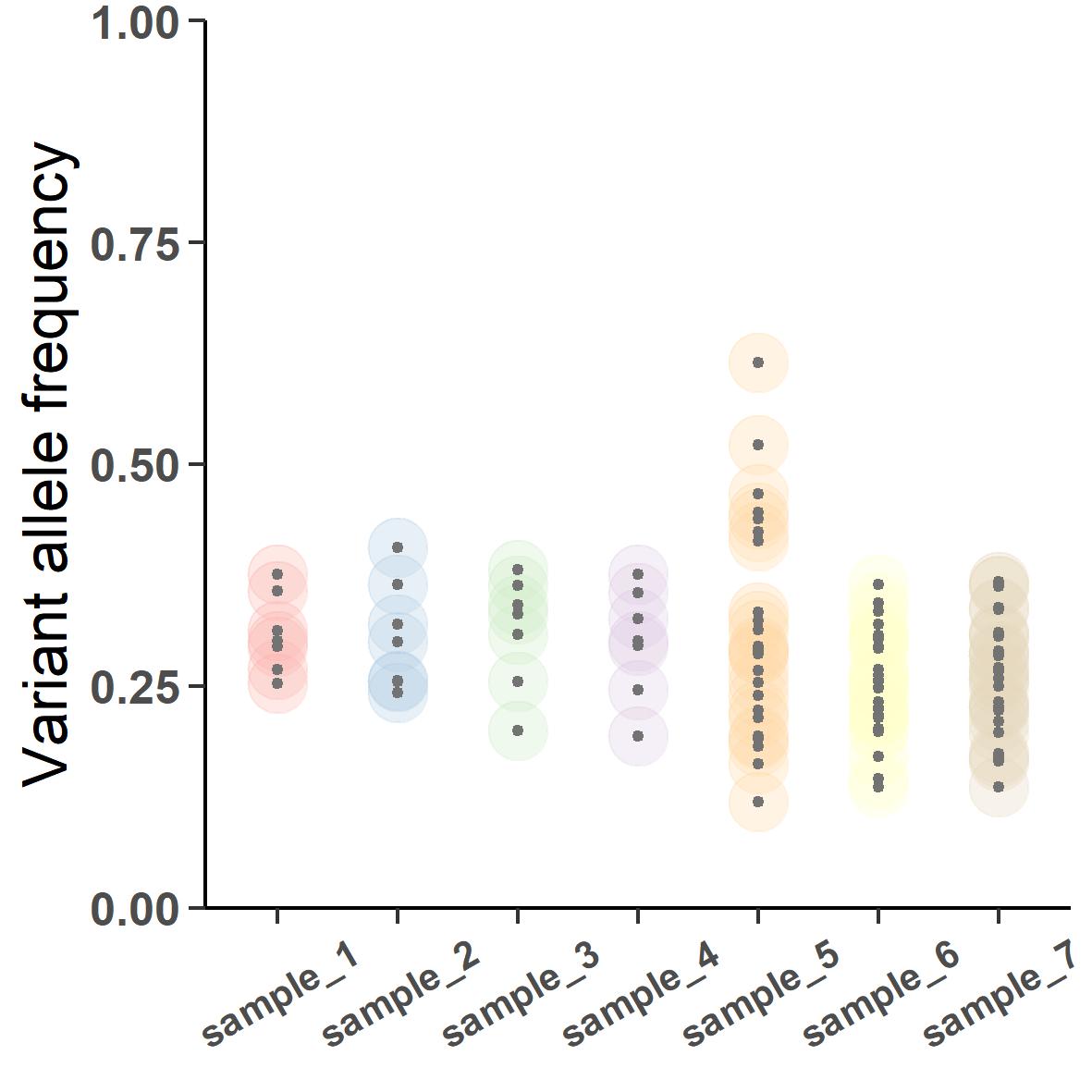
Case in point: a paediatric neuroblastoma tumor is sampled seven times over the duration of the disease. At presentation when biopsy was collected, the tumor tissue had a lot of normal cells nearby that were also sampled. Later the disease metastasized which was bipsied with a much higher content of the metastatic cells.
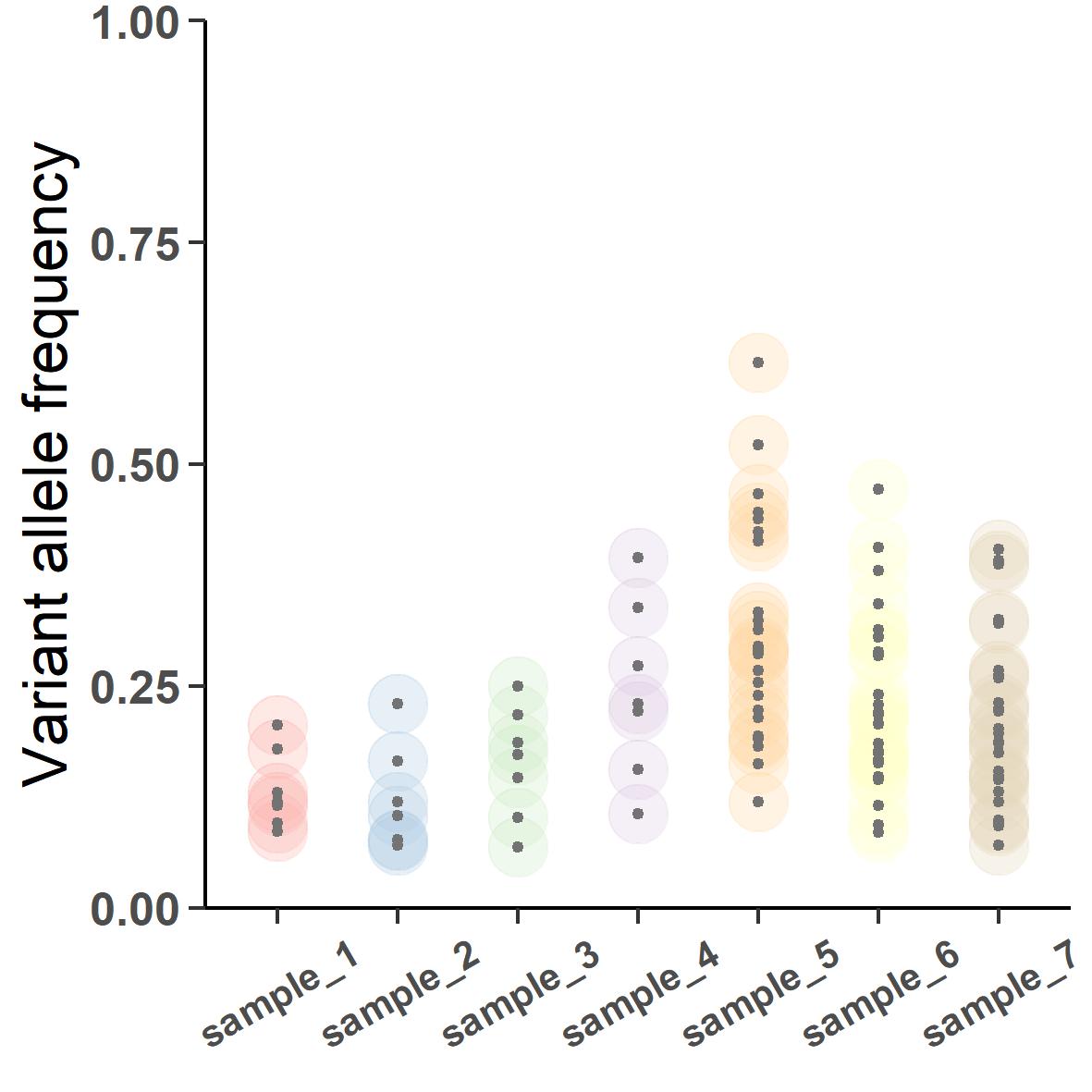
Figure 4 shows the seven tumor samples biopsied from the patient. sample_1 to 3 are from primary tumor and sample_4 to 7 are from the metastasis. Notice the changing dispersion of the VAFs. The primary samples are collected at around 30% purity whereas the metastatic samples were collected during portmortem resulting in a much higher purity of about 90%. If we were to subclonally deconstruct this tumor, the output would look like this:
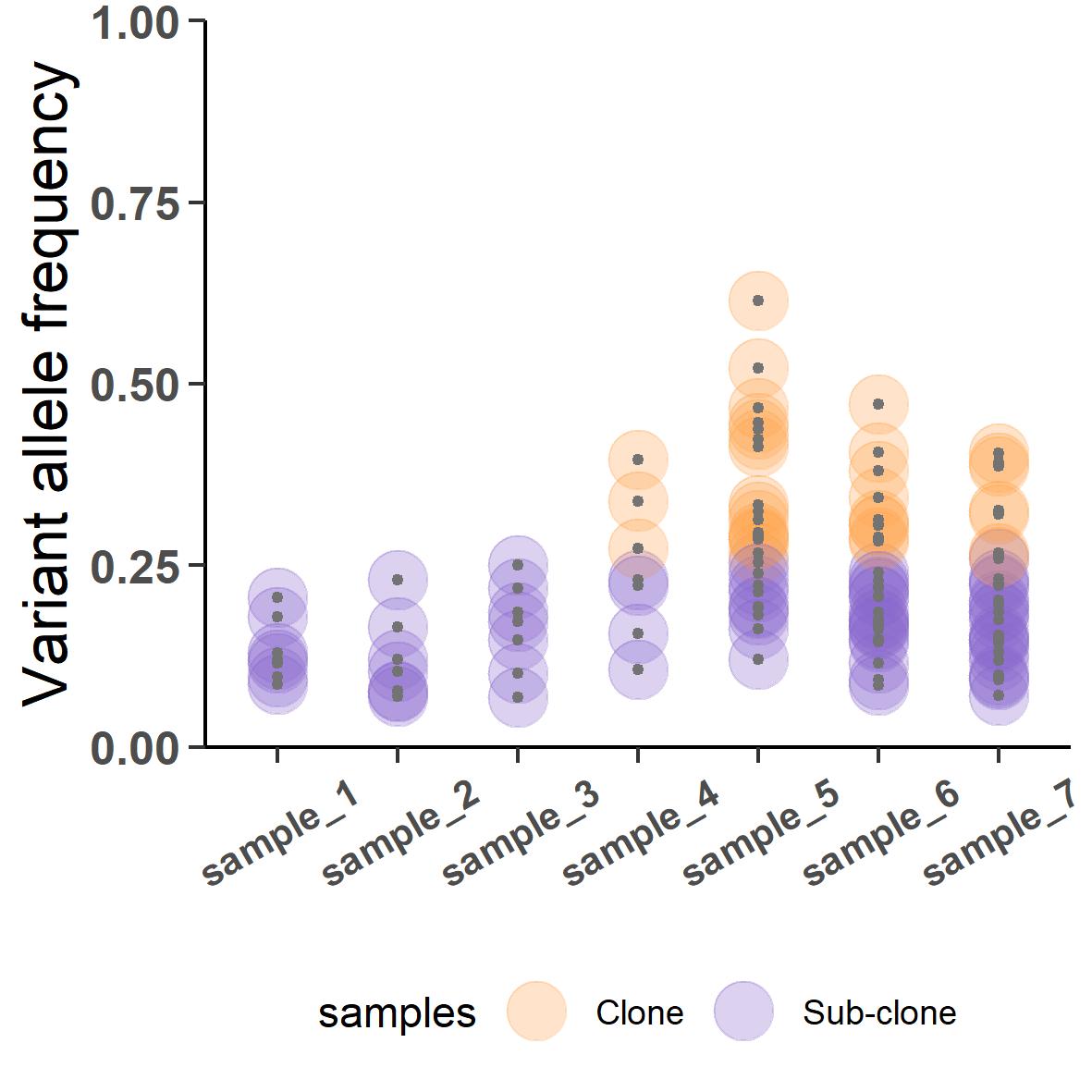
But this will be a contentious inference where all variants in the primary samples are predicted to be subclonal and some of them are apparently jumping ship to fixate clonally in the metastatic samples. The analysis went wrong because we did not take into account the varied purity of the samples. CRUST can normalize this VAFs according to their respective purity which if we were to deconvolve will result in something much closer to the truth:
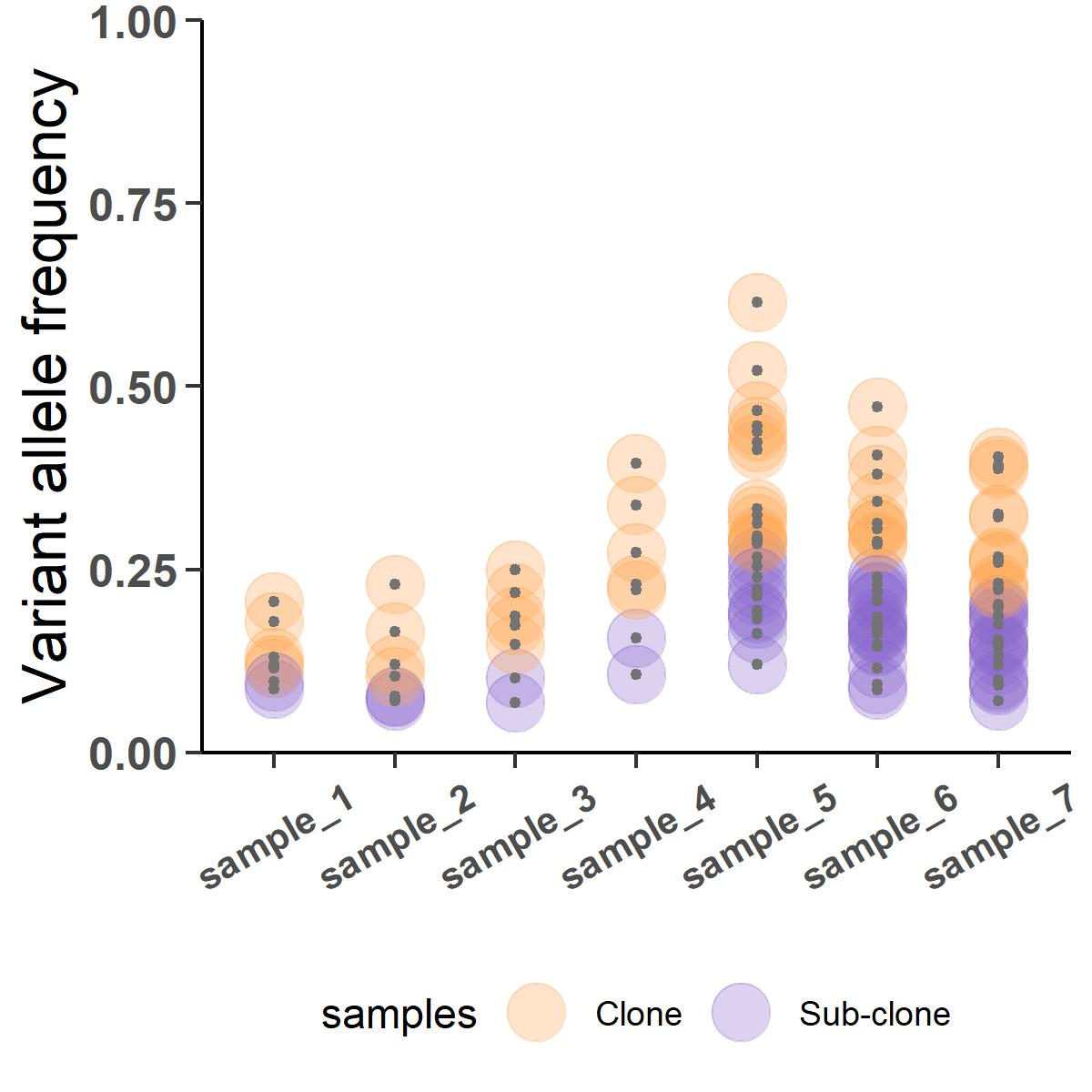
Subjective post-hoc input
CRUST also allows user to re-analyze specific samples based on a brute force suggestion provided by a user. Usually this function would not be needed if allelic compositions are all same for the variants analyzed. But knowing the cost and scope of bulk array sequencing, it is not unconcievable that from time to time allele specific segmental copy number data may not be available to a user. In such cases, if one harbors uncharacteristically strong suspecion that one or more samples are in altered copy number state, the cluster.doubt function provided a unique leeway to remeday that by deconvolving said samples according a user defined clustering input.
Advisory: We recommend not using this function unless you know exactly what you are doing.
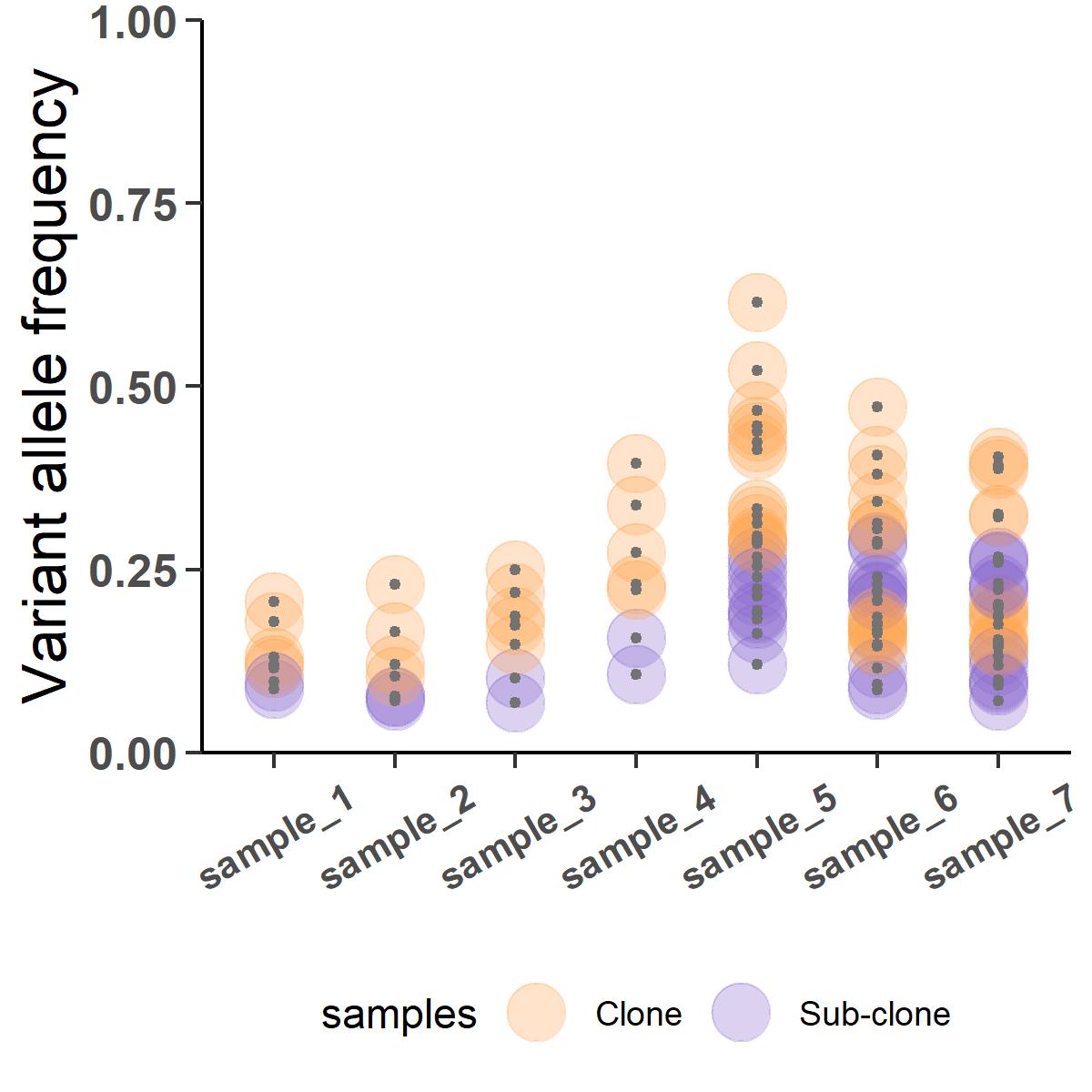
Figure 8 cluster.doubt is invoked to subjectively deconvolve the last two metastatic samples into 4 clusters (2 clones and 2 subclones).
Estimation of allelic composition
We saw how if segmental copynumber data is unavailable to the user the results can be modified to reflect suspected alterations in the copynumber profiles but this can be also rigorously estimated with CRUST. If the sequencing summaries (read depth summaries) for the constitutional genome is available, the AlleleComp function estimates the segmental copynumber for each sample and each chromosome separately. For this the program first starts by reading in .vcf files of a single sample. Multiple samples can also be dynamically read, a small function for this is provided auxiliary scripts
At first the relative coverage estimates are corrected for variation in G/C neucleotide content:
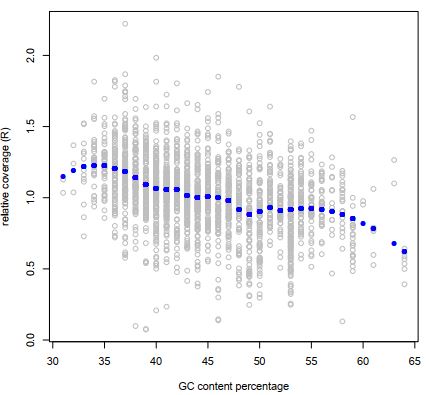
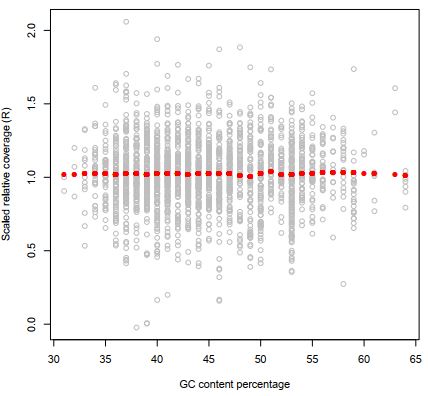
Figure 9 Effect of scaling with GC contenton the relative coverages
Next we get estimates for each chromosome separately that includes segmental allele specific copynumbers. This can be used in conjunction with the sequencing summary to segregate the allels according to allelic makeups and analyze individually.
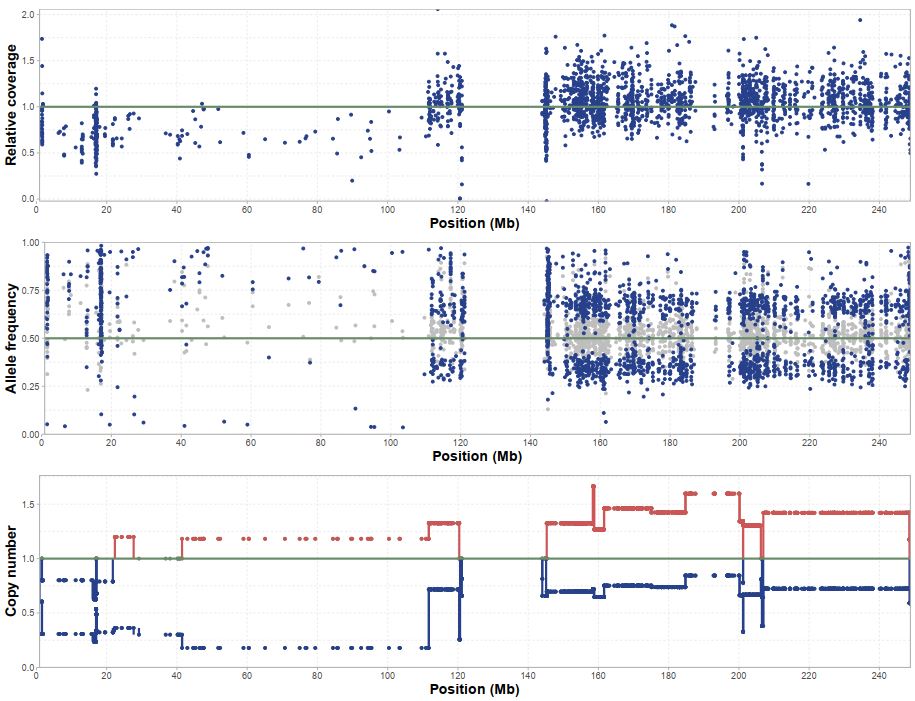
Estimation of allelic imbalance
Along with the copy number estimates CRUST also provides estimates of allelic imbalances. This plotted against negative log transformed relative coverage values can isolate variants chromosomal segments according to thei allelic makeup (example: 0n+2n / 1n+1n / 2n+0n for diploid sample). This plot can aid users in determining the plausible clonality status is is shown here
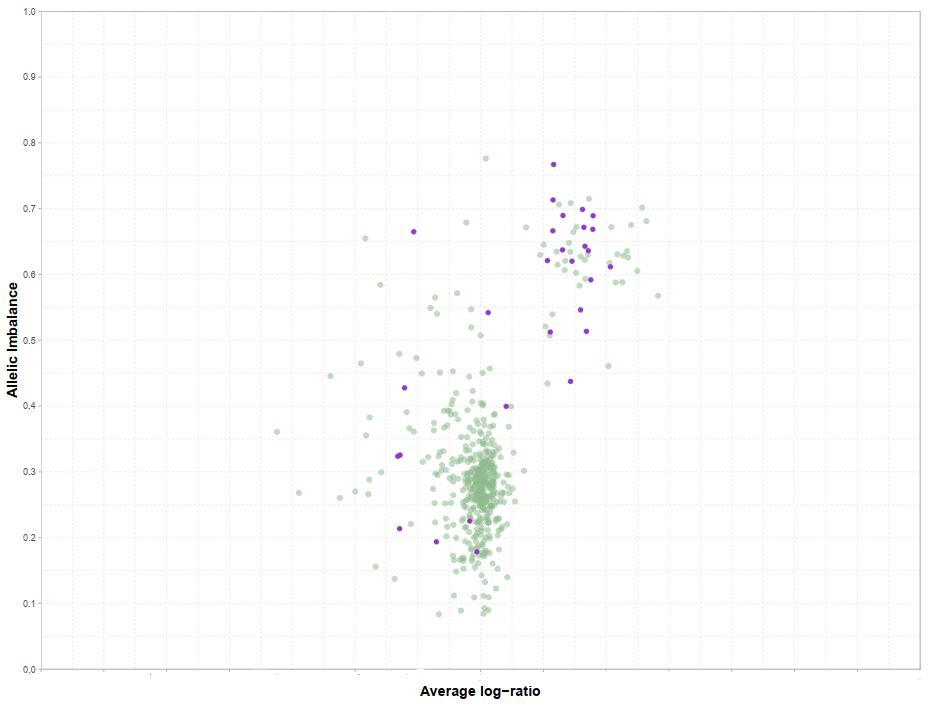
Auxiliary functions
Mutect2 is a popular variant caller that can be used to obtain quality controlled variant calls. Those calls can be tabulated with the GATK extension VariantsToTable. CRUST includes the function mutect2.qc that can convert such an output to a data file compatible with the internal functions.
## Example whole exome seq data is provided in this repository
## Let's query the function to be used:
?mutect2.qc
## A test data is provided with the package
WES <- readr::read_table2("WES.tsv")
sample.name <- c("sample1","sample2","sample3")
CS.dat <- mutect2.qc(WES,sample.name)
A test is provided to check the goodness of the cluster fit. This will indicate existence of outliers. For this the output to the cluster.doc or cluster.doubt is used as input.
## Possible outlying variants are shown
T.goodness.test(r.es)$rej
Coming soon
Functions to import sequencing summary and copy number data from popular third party tools.
More features will be added gradually. If you’d like to see a specific feature incorporated in CRUST consider sending a request here